国内首个真实世界证据支持药物研发与审评的指导文件发布
文件旨在厘清药物研发和监管决策中真实世界证据的相关定义,指导真实世界数据收集以及适用性评估,明确真实世界证据在药物监管决策中的地位和适用范围,探究真实世界证据的评价原则,为工业界和监管部门利用真实世界证据支持药物监管决策提供参考意见。

业界和学界都对其评价颇高。有学者认为这是一个里程碑式的事件,因为它是从监管理念上的极大的转变,真实世界研究适应了当前科学发展的方向,将会是药品研发的一个重要方向。
早在去年5月征求意见稿发布时,在医药界已经引起很大的震动。新药研发耗时长、成本高,从而造成患者负担重,这是世界难题,在降低药物研发成本方面,真实世界研究可谓是为数不多的创新方向之一。
药企非常欢迎真实世界证据,一家跨国药企的管理人员认为,之前已有很多药品通过真实世界数据获批新适应证,以前可能是实在没有办法的情况下,会考虑真实世界数据,有了政策之后,会更方便,或更早期去讨论使用真实世界证据。

《原则》明确,真实世界证据是指通过对适用的真实世界数据进行恰当和充分的分析所获得的关于药物的使用情况和潜在获益-风险的临床证据,包括通过对回顾性或前瞻性观察性研究或者实用临床试验等干预性研究获得的证据。
真实世界数据的常见来源包括但不限于:卫生信息系统(HospitalInformation System,HIS),医保系统,疾病登记系统,国家药品不良反应监测哨点联盟,自然人群队列和专病队列数据库,组学相关数据库,死亡登记数据库,患者报告结局数据,来自移动设备端的数据等。
2016年12月美国通过的《21世纪治愈法案》,通常被视为真实世界证据支持药物和其他医疗产品的监管决策、加快医药产品研发的起点。在这之后,RWS已成为全球相关监管机构、制药工业界和学术界共同关注且极具挑战性的问题。
国家药监局此次《指导原则》的发布,仅仅比美国晚了3年,我国在新药审评审批上的创新越来越快了。
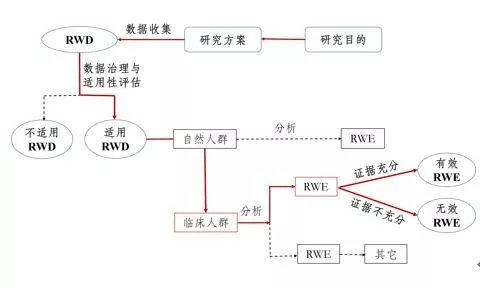
《原则》指出,真实世界研究是指针对预设的临床问题,在真实世界环境下收集与研究对象健康有关的数据(真实世界数据)或基于这些数据衍生的汇总数据,通过分析,获得药物的使用情况及潜在获益-风险的临床证据(真实世界证据)的研究过程(如图1所示)。

图1支持药物监管决策的真实世界研究路径(实线所示)
真实世界证据支持药物监管决策的主要应用范围
真实世界证据应用于支持药物监管决策,涵盖上市前临床研发以及上市后再评价等多个环节。例如,为新产品批准上市提供有效性或安全性的证据;为已获批产品修改说明书提供证据,包括增加或修改适应证,改变剂量、给药方案或给药途径,增加新适用人群,增加实效比较信息,增加安全性信息等;作为上市后要求的一部分支持监管决策的证据等。
罕见病药物准入、广谱药物(例如PD-1抑制剂)新适应证审批、中医药再评价,被认为是最快能从《指导原则》中获益的方向。
真实世界证据支持药物监管决策的主要应用范围有:
1.为新药注册上市提供有效性和安全性的证据
中国有2000万左右罕见患者。2018年5月,国家发布《第一批罕见病目录》,纳入121种罕见病,大约影响300万名患者。截至2018年12月,其中74种在美国或欧盟、日本等地有162种治疗药品。当中只有83种在中国上市,可治疗53种罕见病。
每一种罕见病药物,几乎就代表了一个群体的生存希望。但有一半国外已获批的罕见病药物未能进入中国,很重要的原因就在于临床试验很难完成。
传统的临床试验需要一个实验组和一个对照组,罕见病人本来就很稀少,想要招募到足够的病人入组就成了最大的挑战。
但如果能用真实世界数据来展示疾病的自然特征,将它和实验组进行对比,就可以少招募一半的病人,这无疑会大大减轻药企的成本,加快罕见病新药在国内的注册上市。
另外,对适应证很广谱的药物,比如像PD-1抑制剂,新药上市之后,通常有很多超适应证用药的情况,怎么去迅速扩展更多适应证,真实世界研究是非常好的一种方法。
2.为已上市药物的说明书变更提供证据
对于已经上市的药物,新增适应证通常情况下需要RCT支持。但当RCT不可行或非最优的研究设计时,采用PCT或观察性研究等生成的真实世界证据支持新增适应症可能更具可行性和合理性。
增加或者修改适应证;改变剂量、给药方案或者用药途径;增加新的适用人群;添加实效比较研究的结果;增加安全性信息;说明书的其他修改。
3.为药物上市后要求或再评价提供证据
基于RCT证据获批的药物,通常由于病例数较少、研究时间较短、试验对象入组条件严格、干预标准化等原因,存在安全性信息有限、疗效结论外推不确定、用药方案未必最优、经济学效益缺乏等不足,需要利用真实世界数据对药物在真实医疗实践中的效果、安全性、使用情况,以及经济学效益等方面进行更全面的评估,并不断根据真实世界证据做出决策调整。
4.名老中医经验方、中药医疗机构制剂的人用经验总结与临床研发
对于名老中医经验方、中药医疗机构制剂等已有人用经验药物的临床研发,在处方固定、生产工艺路线基本成型的基础上,可尝试将真实世界研究与随机临床试验相结合,探索临床研发的新路径。
5.真实世界证据用于监管决策的其他应用,包括指导临床研究设计和精准定位目标人群等。
真实世界研究的意义和价值
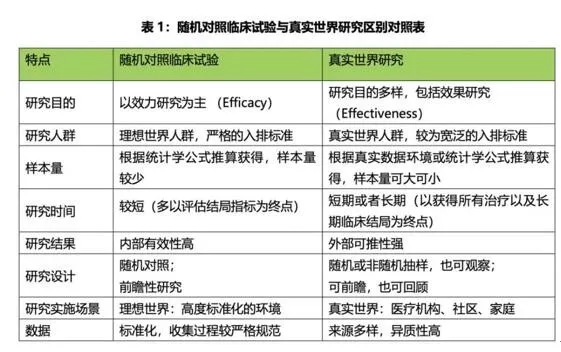
相对于传统的药物临床试验的随机对照试验(RandomizedControlled Trial,RCT)方法,在评价药物安全性和有效性时常常会带来时间久、成本高的代价,如业内共识一款新药平均研发10年、成本10亿美金的说法,最近几年,这个成本还在上升。
而真实世界研究,不仅可以利用真实世界数据支持审评,可以部分取代目前药物研发中的临床试验,节省药物研发成本。
此外,真实世界数据有些是前瞻性的,也有一些是回顾性的,基于历史数据进行注册审批,还能缩减药品审批上市的时间,因此受到药企欢迎。

RCT与RWE对比(来源:《真实世界研究指南2018版》)
基于此,《原则》中也列举了两项真实世界证据的研究案例:
示例1:利用真实世界证据支持新增适应证
申办方在某药上市后发起一项通过真实世界数据评价其在中国女性中减少临床骨质疏松性骨折的有效性和安全性研究。
该研究遵循真实世界研究的良好实践,研究方案事先公开。真实世界数据来源具有良好的研究人群代表性,样本量达4万余人。
该研究的主要终点通过病历审查进行验证,以倾向评分匹配作为主要分析方法,同时使用逆概率加权法、高维倾向评分调整等多种方法进行敏感性分析,并定量评估未测量到的混杂因素的影响。
该真实世界研究的结果与全球RCT研究相近,并用不同数据来源、不同研究机构的真实世界数据重现出该结果。

示例2:利用真实世界证据支持扩大联合用药

真实世界中患者所联合的化疗方案并不局限于卡铂与紫杉醇,还包括培美曲塞联合铂类、吉西他滨联合顺铂等。2018年10月该药获批将治疗方案扩展为联合以铂类为基础的化疗方案,其中三项真实世界研究结果提供了强有力的支持证据。
这三项研究回顾性分析了山东省肿瘤医院、江苏省肿瘤医院和中国医学科学院肿瘤医院三家医院的真实世界患者数据,均显示在含铂双药化疗基础上联合贝伐珠单抗较单纯化疗显著延长PFS和OS,与全球人群数据具有一致性,并且未发现新的安全性问题。
此外,相关真实世界研究还提供了EGFR突变和脑转移等不同患者亚组中的疗效数据,从多角度证实了贝伐珠单抗联合疗法的有效性和安全性。
真实世界数据指导细则正在研讨推进
真实世界证据对药物研发来说,最直接的价值就是降低成本。新药研发时间长、成本高、成功率低,多年前就有一款新药平均研发10年、成本10亿美金的说法,最近几年成本还在上升。

这也是药企欢迎真实世界证据的原因之一,前述跨国药企的管理人员说,对于新药研发,真实世界证据可以在一定程度上减少Ⅲ期临床试验的成本;对于扩适应证的药品,可以直接免去Ⅲ期临床试验。

有专业人士认为,对于符合真实世界证据要求的企业来讲,它的成本和时间将会急剧的降低,尤其是广谱药拓适应证,用真实世界证据非常好,不过随机对照临床试验还会是新药上市的主流,真实世界证据作为辅助和补充。
在政策初期,药企如果可以负担随机对照临床研究,可能还是会优先选择随机对照临床研究,毕竟真实世界证据还需要不断进一步验证。政策实施之后,会有很多的企业会去尝试发起真实世界研究,但最终能真正有效可靠的证据不会太多,这是因为作为真实世界研究前提的真实世界数据基础还不够。

真实世界研究,有些是前瞻性的,也有一些是回顾性的,基于历史数据进行注册审批。基于历史数据的研究并不完全取决于企业本身是否有能力,更重要的是目前数据的可获得性,以及它们的质量能否满足注册的需要。
过去,医疗系统内的数据本就不是为了注册需要,主要是出于监管的需要,比如医保系统首要是为了满足医保作为支付监管的任务,但真实世界研究需要注册申请、评估效果、说明产品有效性、安全性的数据,原有系统的数据丰富程度还不够。
不过,《指导原则》中大篇幅地提出了对于真实世界数据的表述,包括数据相关性和可靠性的评估以及统计方法和研究方法的介绍,说明监管部门在政策的制定上非常落地。
如果说《指导原则》的出台是为真实世界证据的应用确立了规范,那么接下来政策的顺利落地还将需要更多的细则和标准。据了解,关于真实世界数据的指导细则也研讨推进中,规定什么样的真实世界数据可以获得真实世界证据,进而拓展适应证。
最新新闻




北京含元资本管理有限公司
专注于双生(生物科技和生命健康)、双智(智能制造和人工智能)、双碳(新能源和新材料)、双新(新时代和新生代下的新消费)领域的投资机构。
关注我们
